Abstract: Rhodium acetate, related rhodium carboxylates, and rhodium amide complexes are powerful catalysts for carbene chemistry. They readily promote the decomposition of diazo compounds and transfer the resulting carbene to a variety of substrates. There have been several quantum chemistry studies of these compounds, particularly the acetate. These have all used non-relativistic methods, and all have shown optimized Rh-Rh bond lengths significantly longer than the experimental value. In this study we have surveyed several scalar relativistic DFT methods using both Gaussian and numerical basis functions (in DGAUSS and DMOL3). Several combinations of exchange-correlation functionals with relativistic and non-relativistic effective core potentials (ECP) were investigated, as were non-relativistic and all electron scalar relativistic methods. The combination of the PW91 exchange and PW91 correlation functional with the Christiansen-Ermler ECP gave the best results: 2.3924 Å compared to the experimental value of 2.3855 Å.
Note: this web page makes some use of chime to display molecular structures.
Introduction: The chemistry of rhodium acetate and related
compounds as catalysts for carbene chemistry has been well established.1
This paper is concerned mostly with the diaquo complex of rhodium acetate,
shown below. If you see a blank window, drag your cursor over the
window and the structure should appear, in you have chime installed.
If not, please see the link above.
In this study we present the results of a comparison of DFT methods for computing the structure of the diaquo complex of rhodium acetate. We compare non-relativistic calculations to calculations that include scalar relativistic effects either via effective core potentials, or all electron methods; in addition we compare results from Gaussian and numerical basis sets. In the calculations described here, all of the relativistic calculations were scalar in nature: this means that no attempt was made to solve the complete Dirac equation with four-component wavefunctions, and spin-orbit coupling was not explicitly included. Thus one-component wavefunctions were used, and only the mass-velocity and Darwin terms in the relativistic Hamiltonian were retained. Faegri8 has shown that including these scalar effects reproduces most of the relativistic bond contraction seen in fully relativistic four-component calculations.
Computations: The computations described here were done using DFT methods with either DGAUSS or DMOL3. DGAUSS uses Gaussian basis sets that have been optimized for solving the Kohn-Sham equations. All of the DGAUSS calculations were done with an all electron double zeta valence polarized (dzvp) basis set, except that in some cases, 28 core electrons on the Rh atoms were replaced with an effective core potential (ECP), often one that was derived from solving the Dirac-Fock equation. Such a potential is referred to as a relativistic effective core potential (RECP). DMOL3 uses a numerical basis set; the calculations here used one such set (DNP) that is comparable in quality to the dzvp basis set in DGAUSS. DMOL3 has a facility that applies scalar relativistic corrections to this basis set;9 this method was used for the relativistic calculations in DMOL3. Geometry optimizations in DGAUSS were done with the medium geometry convergence and medium scf convergence criteria; a few test calculations were done with both of these set to tight. In these tests, the resulting change in the Rh-Rh bond length was very small, and was never more than 0.001 Å. In all cases the high accuracy option for the numerical integrals was set. The DMOL3 calculations were done with either 1x10-6 scf energy combined with 1x10-5 geometry convergence criteria, or 1x10-8 scf energy combined with 1x10-6 geometry convergence criteria. These tighter criteria were used in an attempt to improve the vibrational frequencies calculated with DMOL3; as in DGAUSS they had an insignificant effect on the Rh-Rh bond length. In both programs the exchange-correlation potential was included in a self-consistent fashion in all calculations. All DGAUSS calculations were run on a Silicon Graphics Origin 2000 computer using either 4 or 8 processors; all of the DMOL3 calculations were done on a parallel IBM SP computer, usually on 20 processors, except for the calculations with the PBE functional which were done on the SGI Origin.
The DGAUSS calculations used the following ECP's: Stevens-Krauss10, Christiansen-Ermler11, Hay-Wadt12, Stuttgart13, and DGAUSS14. The scalar relativistic method in DMOL3 is due to Delley9. While most of the functionals in this study are in common use, the BOP15 and PBE16 functionals in DMOL3 are not. It should be noted that DMOL3 refers to the PW91-PW91 functional as GGA.
Results and Discussion: The bond lengths predicted by the
various methods surveyed are shown in the tables below. The experimental
value is 2.3855 Å2. For the calculations with DMOL3
we found the following Rh-Rh bond lengths in Å.
| Functional | PBE | GGA | BOP | BLYP |
| Relativistic Bond Length | 2.411 | 2.409 | 2.439 | 2.438 |
| Non-Relativistic Bond Length | 2.421 | 2.42 | 2.451 | 2.449 |
For the DGAUSS calculations we found the following bond lengths.
The best agreement with the X-ray crystal structure is shown in red.
| Relativistic | BP | BLYP | B88-PW91 | PW91-PW91 | ||
| Stevens-Krauss | 2.42297 | 2.44306 | 2.42269 | 2.42022 | ||
| Christiansen-Ermler | 2.39301 | 2.41299 | 2.39302 | 2.3918 | ||
| Hay-Wadt | 2.42046 | 2.44034 | 2.42047 | 2.41926 | ||
| Stuttgart | 2.41479 | 2.436 | 2.41522 | 2.41506 | ||
| DGAUSS | 2.47109 | 2.48804 | 2.46567 | 2.46586 | ||
| All Electron (no ECP) | 2.4184 | 2.43903 | 2.41692 | 2.41632* | ||
These results all show that in DMOL3 the all electron scalar relativistic corrections produces a contraction of the Rh-Rh bond for all of the functionals considered. In DGAUSS, all of the relativistic ECP's produce also show a contraction of this bond, when compared to a similar non-relativistic ECP, but they do not always show similar contraction compared to a non-relativistic all-electron calculation. The Christiansen-Ermler RECP shows the greatest contraction and comes closest to reproducing the experimental bond length when it is used with the PW91-PW91 functional. We interpret these results as showing that the Rh-Rh bond length in diaquo rhodium acetate is at least in part determined by relativistic effects, and that of the methods surveyed, the combination of the PW91-PW91 functional with the Christiansen-Ermler RECP is the best at reproducing these effects. As mentioned previously the above DGAUSS results were obtained with somewhat relaxed convergence criteria; when the most stringent criteria were used, this best method gave 2.3924 Å, a difference of less than 0.001 Å.
In order to check the apparent accuracy of this method, the Christiansen-Ermler RECP was used with PW91-PW91 to predict the Rh-Rh bond length in diaquo rhodium trifluoroacetate; the result was 2.4156 Å, which agrees fairly well with the experimental18 value of 2.409 Å. A survey of the Cambridge Crystallography database which found 59 structures containing the rhodium trifluoroacetate unit gave an average Rh-Rh bond length of 2.414 Å with a standard deviation of 0.018 Å; the calculation is in excellent agreement with this average value.
In some cases the geometry optimizations did not converge to equilibrium structures, instead structures with one (or in a few cases more then one) imaginary vibrational frequencies were found. In most cases when this happened, the imaginary frequencies were eliminated by re-optimizing the structure guided by the Hessian from the frequency calculation. In a few cases, this process was not successful. The imaginary frequency (frequencies) were still present. The vibrational modes for these were always hindered rotational modes for one or more of the methyl groups. This is illustrated by the following movie (a screen capture from the UNICHEM interface to DGAUSS; caution: the movie is about 2Mb in size). Less smooth animation of the corresponding vibrational mode in rhodium trifluoroacetate is shown in this chime link. If you do not see the animation, right click in the chime window and select animation.
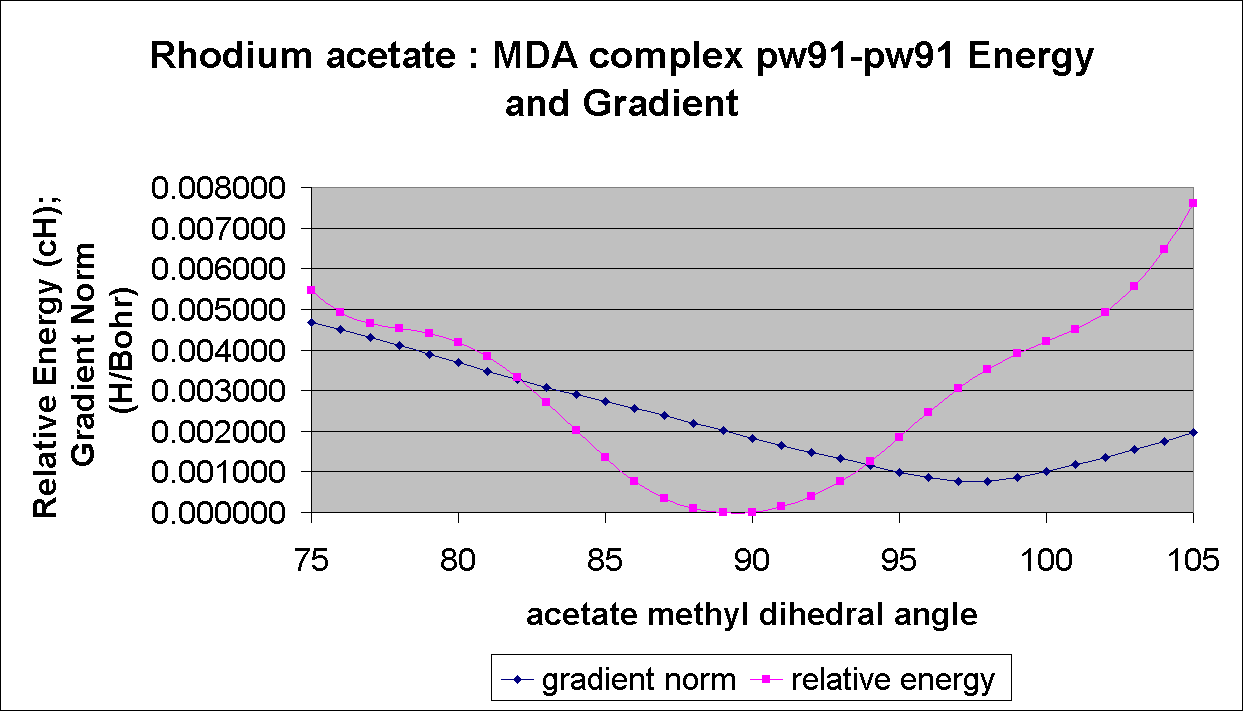
This problem is not too serious for this study, since the Rh-Rh bond is not involved in any of the imaginary frequency vibrational modes. However, we are conducting a parallel study of relativistic effects in the catalytic chemistry of rhodium acetate. This study involves finding transition states for rhodium acetate catalyzed diazo decompositions, and unwanted imaginary frequencies are a significant problem here. Our (incomplete) results in this area show barriers that differ from experiment by a few kcal/mol. A single low frequency that is improperly found to be imaginary causes an error of about 0.5 kcal/mol in the barrier at room temperature. Thus several of these errors can completely obscure meaningful differences between functionals and ECP's. The PW91-PW91 functional in DGAUSS serves to illustrate this problem. It seems that for the PW91-PW91 functional in DGAUSS the geometry with a minimum in the gradient does not correspond to the nearest geometry with a minimum in the energy, as shown here for a scan of an acetate methyl dihedral angle in the complex formed between rhodium acetate and methyl diazoacetate (MDA).
This problem of spurious vibrational frequencies in DGAUSS that are due to poor alignment of the gradient and energy minima is serious since it is the gradient that drives the geometry optimization. Any optimization that suffers from this poor alignment will find a geometry that is a gradient minimum and not an energy minimum. There does not seem to be a way in DGAUSS to minimize on the energy only during an optimization. In DMOL3 there are separate convergence criteria for these and the user can switch one or the other off, if desired. The spurious imaginary frequencies in DMOL3 optimizations seem to be related to the step size used for the numerical energy second derivatives. This problem is being studied now.
Conclusions: Of the methods surveyed, the combination of the Christiansen-Ermler ECP with the PW91-PW91 functional in DGAUSS did a better job of predicting the Rh-Rh bond length in rhodium acetate, and this ECP combined with most functionals was more accurate than most of the other methods.
Acknowledgment: This work was supported by the North Carolina Supercomputing Center.
References:
1) Modern Catalytic Methods for Organic Synthesis with Diazo Compounds
from Cyclopropanes to Ylides, Doyle, M. P.; McKervey, M. A.; Ye, T.
John Wiley & Sons, New York, 1998.
2) Cotton, F. A.; DeBoer, B. G.; Laprade, M. D.; Pipal, J. R.; Ucko,
D. A. Acta Crystallogr. 1971, B27, 1664.
3) Norman, J. G. Jr.; Kolari, H. J. J. Amer. Chem. Soc.1978,
100,
791; Nakatsuji, H.; Onishi, Y.; Ushio, J.; Yonezawa, T. Inorg.
Chem. 1983, 22, 1623-1630; Mougenot, P.; Demuynck, J.;
Benard, M. Chem. Phys. Lett. 1987, 136, 279-282; Nakatsuji,
H.; Ushio, J.; Kanda, K.; Onishi, Y.; Kawamura, T.; Yonezawa, T. Chem.
Phys. Lett. 1981, 79, 299-304; Rodriguez-Garcia, C.; Gonzalez-Blanco,
O.; Oliva, A.; Ortuno, R. M.; Branchadell, V. Eur. J. Inog. Chem.2000,
5,
1073-1078; Doyle, M. P.; Winchester, W. R.; Hoorn, J. A. A.; Lynch,
V.; Simonsen, S. H.; Ghosh, R. J. Amer. Chem. Soc. 1993,
125,
9968-9978; Padwa, A.; Snyder, J. P.; Curtis, E. A.; Sheehan, S. M.; Worsencroft,
K. J.; Kappe, C. O. J. Amer. Chem. Soc. 2000,
122,
8155-8167; Morehead, A. T. Jr. Mechanistic Investigation of the
Dirhodium(II) Catalyzed Decomposition of Diazocarbonyl Compounds, Ph.
D. dissertation, Duke University, Durham, NC, 1996.
4) Cotton, F. A.; Feng, X. J. Amer. Chem. Soc. 1998,
120,
3387.
5) Pyykko, P. Chem. Rev. 1988, 88, 563-594; Relativistic
Effects in Chemistry, Part A Theory and Techniques, Balasubramanian,
K. John Wiley and Sons, New York, 1997.
6) Desclaux, J. P. Atomic Data and Nuclear Data Tables 1973,
12,
311.
7) Diefenbach, A.; Bickelhaupt, F. M. J. Chem. Phys. 2001,
115,
4030.
8) Faegri, K. Jr.; Saue, T. J. Chem. Phys. 2001, 115,
2456-2464.
9) Delley, B. Int. J. Quantum. Chem. 1998, 69,
423-433.
10) Stevens, W. J.; Krauss, M.; Basch, H.; Jasien, P. G. Can. J.
Chem., 1992, 70, 612-629.
11) LaJohn, L. A.; Christiansen, P. A.; Ross, R. B.; Atashroo, T.;
Ermler, W. C. J. Chem. Phys. 1987, 87, 2812-2824.
12) Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82,
284-298; Hay, P. J. Wadt, W. R.; J. Chem. Phys. 1985, 82,
299-310.
13) Kuchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Mol. Phys.,
1991,
74,
1245-1263.
14) Chen, H.; Krasowski, M.; Fitzgerald, G.; J. Chem. Phys. 1998,
11,
8710-8717.
15) Tsuneda, T.; Hirao, K.; Chem. Phys. Lett. 1997, 268,
510-520.
16) Perdew, J. P.; Burke, K.; Ernzerhof, M.; Phys. Rev. Lett.
1996,
77, 3865-3868.
17) Gaussian 98, Revision A.11.3, M. J. Frisch, G. W. Trucks, H. B.
Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski,
J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich,
J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas,
J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli,
C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala,
Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari,
J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov,
G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L.
Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara,
M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J.
L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, and J. A.
Pople, Gaussian, Inc., Pittsburgh PA, 2002.
18) Cotton, F. A.; Felthouse, T. R.; Inorg. Chem. 1982,
21,
2667-2675.